The neurobehavioural group have been investigating the mechanisms by which mutations in PER2, a gene known to be involved with human sleep syndromes, can speed up our master body clock by several hours a day.
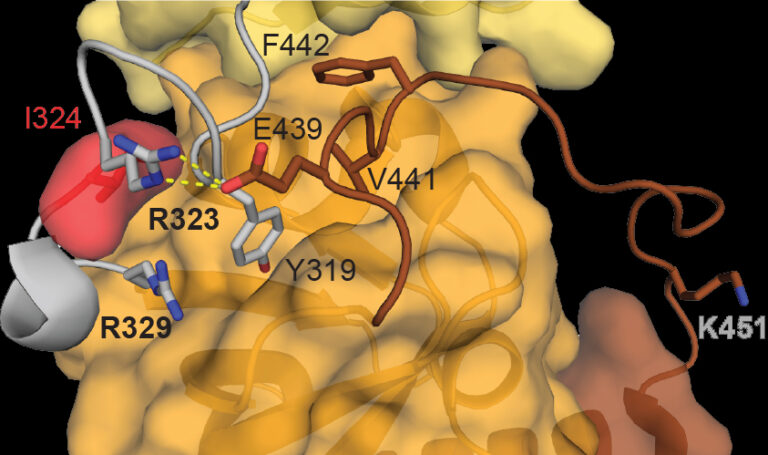
The neurobehavioural group, led by Pat Nolan, have been investigating the mechanisms by which mutations in PER2, a gene known to be involved with human sleep syndromes, can speed up our master body clock by several hours a day A small area in the brain, called the suprachiasmatic nucleus (SCN), is responsible for keeping our bodies in sync with 24 hour time. Within this circadian pacemaker, a complicated network of proteins interact with and co-regulate each other. As the day progresses, the levels of these proteins will be directed to rise and fall; this is how the SCN signals circadian time to the rest of the body. One such protein is called Period2 (PER2), which is encoded by the Per2 gene. PER2 can influence its own production in the SCN: whilst PER2 levels are high, further production of PER2 will be blocked. Gradually over the course of 24 hours, PER2 is degraded and denatured until levels reach a trough. At this point, the SCN will produce more PER2 and the cycle will repeat itself. This timely production and degradation of PER2, along with several other clock proteins such as Cryptochrome (CRY), Period1 (PER1) and Bmal, helps to maintain a daily 24 hour cycle. Mutations in PER2 can cause an inherited sleep disorder called “Familial advanced sleep phase syndrome” (FASPS), as shown by Kong L. Toh et al in 2001. Humans carrying this mutated gene have an accelerated body clock, meaning their daily circadian cycle is accelerated and they display an unusual sleep pattern. In work now published in PNAS, the Harwell neurobehaviour group, in collaboration with Hastings group in MRC LMB and the Partch group in University of California, Santa Cruz, used ENU mutagenesis to introduce a new mutation, which they named “Early doors” (Edo), in the Per2 gene of mice, and found that the circadian cycle of the Edo mutants was an hour and a half shorter than mice with the normal, wild-type, Per2 gene. The group then used a variety of methods to investigate the nature of the Edo mutation further. They found that the mutated PER2 protein was able to carry out its usual function but, each day, it degraded faster in the SCN of Edo mice than in wild-type mice, resulting in a shorter circadian cycle. Usually, the PER2 protein can form a dimer (a pair of proteins joined together) with other clock proteins, such as another PER2, or CRY1. These dimers are important features of the circadian timing mechanism in the SCN. The Edo mutation changes the shape of a section of the PER2 protein called the PAS domain, which is the section that binds with other proteins to make dimers. The team found that although the mutated form of PER2 was still able to form dimers with other proteins, the PER2 in these dimers was less stable than usual, which caused them to degrade more quickly. A second mutation in a gene called Casein kinase 1 (CK1) has also been found to cause FASPS. The non-mutated form of the CK1 protein is one of those responsible for the gradual degradation of PER2 in the SCN throughout the day. A mutant form of this protein is able to degrade PER2 much more quickly, resulting in a similarly accelerated circadian cycle. After deducing how the Edo mutation shortened the circadian rhythm by itself, the team produced mice with mutations in both the Per2 and the Ck1 genes. These double-mutant mice had an ultra-short circadian cycle, more than 5 hours shorter than wild-type mice, and several hours shorter than the Edo mice. Together, these findings emphasise the vital role that PER2 protein stability plays in regulating the SCN rhythm, and keeping our bodies on a 24-hour clock.